CYSTIC FIBROSIS
CF is one of the most common autosomal recessive disorders in individuals of Western European origin, in whom the incidence varies from 1 in 2000 to 1 in 3000. The incidence is slightly lower in Eastern and Southern European populations and much lower in African-Americans (1 in 15 000) and Asian-Americans (1 in 31 000).
CLINICAL FEATURES
The organs most commonly affected in CF are the lungs and the pancreas. Chronic lung disease caused by recurrent infection eventually leads to fibrotic changes in the lungs with secondary cardiac failure, a condition known as cor pulmonale. When this complication occurs the only hope for long term survival rests in a successful heart-lung transplant.
In 85% of persons with CF pancreatic function is impaired with reduced enzyme secretion due to blockage of the pancreatic ducts by inspissated secretions. This leads to malabsorption with an increase in the fat content of the stools. This complication of CF is readily amenable to treatment with oral supplements of pancreatic enzymes.
Other problems which are commonly encountered in CF include nasal polyps, rectal prolapse, cirrhosis and diabetes mellitus. Around 10% of children with CF present in the newborn period with obstruction of the small bowel due to thickened meconium, a condition known as meconium ileus. Almost all males with CF are sterile because of congenital bilateral absence of the vas deferens (CBAVD). It is now recognised that a small subset of males have a very mild form of CF in which CBAVD is the only significant clinical problem. Other rare presentations of CF include chronic pancreatitis, diffuse bronchiectasis and bronchopulmonary allergic aspergillosis.
GENETICS
CF shows autosomal recessive inheritance. The most likely explanation proposed for the high incidence is heterozygote advantage, possibly mediated by increased heterozygote resistance to chloride-secreting bacterial-induced diarrhoea.
The gene was named the CF transmembrane conductance regulator (CFTR) gene and was shown to span a genomic region of approximately 25 kb and to contain 27 exons.
The CFTR protein
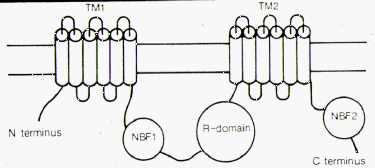
| The structure of CFTR is consistent with a protein product containing 1480 amino acids with a molecular weight of 168kDa. It is believed to consist of two transmembrane (TM) domains which anchor it to the cell membrane, two nucleotide binding folds (NBF) which bind ATP, and a regulatory (R) domain, which is phosphorylated by protein kinase-A. | |
The primary role of the CFTR protein is to act as a chloride channel to reduce the level of intracellular sodium choride, which improves the quality of cellular mucus secretions.
Mutations in the CFTR gene
The first mutation to be identified in CFTR was a deletion of three adjacent base pairs at the 508th codon which results in the loss of a phenylalanine residue. This mutation is known as Delta F508 (Delta for deletion and F for phenylalanine) and it has been shown to account for approximately 70% of all mutations in CFTR with the highest incidence of 88% being in Denmark.
Contribution of Delta F508 mutation to all CF mutations
|
Country |
%
|
|
Denark |
88 |
|
Netherlands |
79 |
|
United Kingdom |
78 |
|
Ireland |
75 |
|
France |
75 |
|
United States |
66 |
|
Germany |
65 |
|
Poland |
55 |
|
Italy |
50 |
|
Turkey |
30 |
Over 800 other mutations in CFTR have been identified. These include missense, frameshift, splicesite, nonsense and deletion mutations. A multiplex PCR technique has been developed which detects approximately 85% of all carriers. Using this it is possible to reduce the carrier risk for a healthy individual from a population risk of 1 in 22 to less than 1 in 140.
Genotype-phenotype correlation
Mutations in CFTR can influence the function of the protein product by:
The net effect of all these mutations is to reduce the normal functional activity of the CFTR protein. The extent to which normal CFTR protein activity is reduced correlates well with the clinical phenotype. Levels of less than 3% are associated with severe 'classic' CF, sometimes referred to as the P1 type because of associated pancreatic insufficiency. Levels of activity between 3% and 8% cause a milder 'atypical' form of CF in which there is respiratory disease but relatively normal pancreatic function. This is referred to as the PS (pancreatic sufficient) form. Finally, levels of activity between 8% and 12% cause the mildest CF phenotype, in which the only clinical abnormality is CBAVD in males.
CLINICAL APPLICATIONS AND FUTURE PROSPECTS
Prior to the mapping of the CF locus and the subsequent isolation of CFTR, it was not possible to offer either carrier detection or reliable prenatal diagnosis. Now parents of an affected child can almost always be offered prenatal diagnosis, either by direct mutational analysis of DNA from chorionic villi, or by linkage analysis using polymorphic intragenic markers if one or both of the mutations in the affected child cannot be identified. Similarly, knowledge of one or both of the mutations in an affected child now permits the offer of carrier detection to close family relatives.